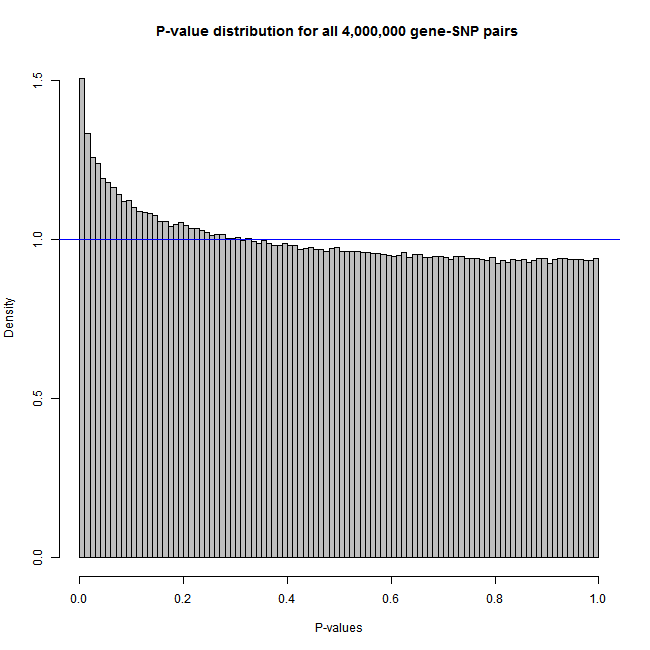
Create an artificial dataset and plot the histogram and Q-Q plot of all p-values
library('MatrixEQTL')
# Number of samples
n = 100;
# Number of variables
ngs = 2000;
# Common signal in all variables (population stratification)
pop = 0.2 * rnorm(n);
# data matrices
snps.mat = matrix(rnorm(n*ngs), ncol = ngs) + pop;
gene.mat = matrix(rnorm(n*ngs), ncol = ngs) + pop + snps.mat*((1:ngs)/ngs)^9/2;
# data objects for Matrix eQTL engine
snps1 = SlicedData$new( t( snps.mat ) );
gene1 = SlicedData$new( t( gene.mat ) );
cvrt1 = SlicedData$new( );
rm(snps.mat, gene.mat)
# Slice data in blocks of 500 variables
snps1$ResliceCombined(500);
gene1$ResliceCombined(500);
# name of temporary output file
filename = tempfile();
# Perform analysis recording information for
# a histogram
meh = Matrix_eQTL_engine(
snps = snps1,
gene = gene1,
cvrt = cvrt1,
output_file_name = filename,
pvOutputThreshold = 1e-100,
useModel = modelLINEAR,
errorCovariance = numeric(),
verbose = TRUE,
pvalue.hist = 100);
unlink( filename );
# png(filename = "histogram.png", width = 650, height = 650)
plot(meh, col="grey")
# dev.off();
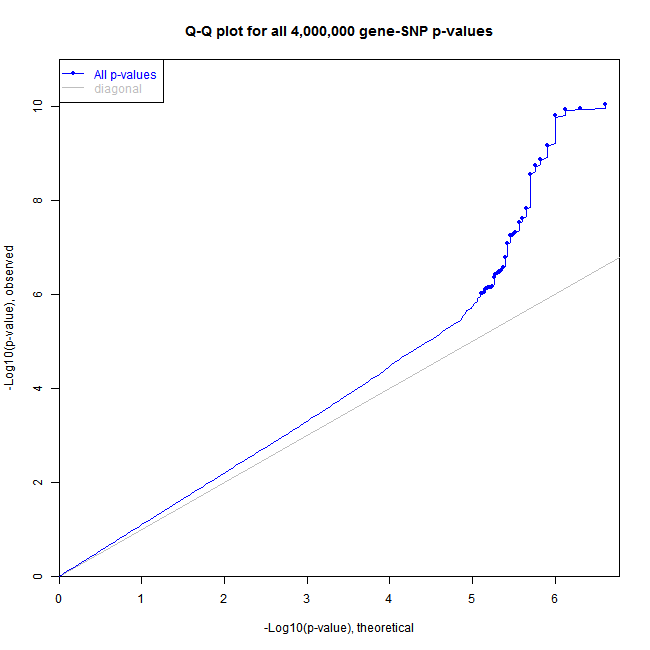
# Perform the same analysis recording information for
# a Q-Q plot
meq = Matrix_eQTL_engine(
snps = snps1,
gene = gene1,
cvrt = cvrt1,
output_file_name = filename,
pvOutputThreshold = 1e-6,
useModel = modelLINEAR,
errorCovariance = numeric(),
verbose = TRUE,
pvalue.hist = "qqplot");
unlink( filename );
# png(filename = "QQplot.png", width = 650, height = 650)
plot(meq, pch = 16, cex = 0.7)
# dev.off();